Automated Reconstruction of Whole-Genome Phylogenies from Short-Sequence Reads
Posted in Publications, publishedNo Comment
F Bertels, OK Silander, M Pachkov, PB Rainey, E van Nimwegen
Automated Reconstruction of Whole-Genome Phylogenies from Short-Sequence Reads
Molecular biology and evolution 2014 31 (5), 1077-1088
ABSTRACT
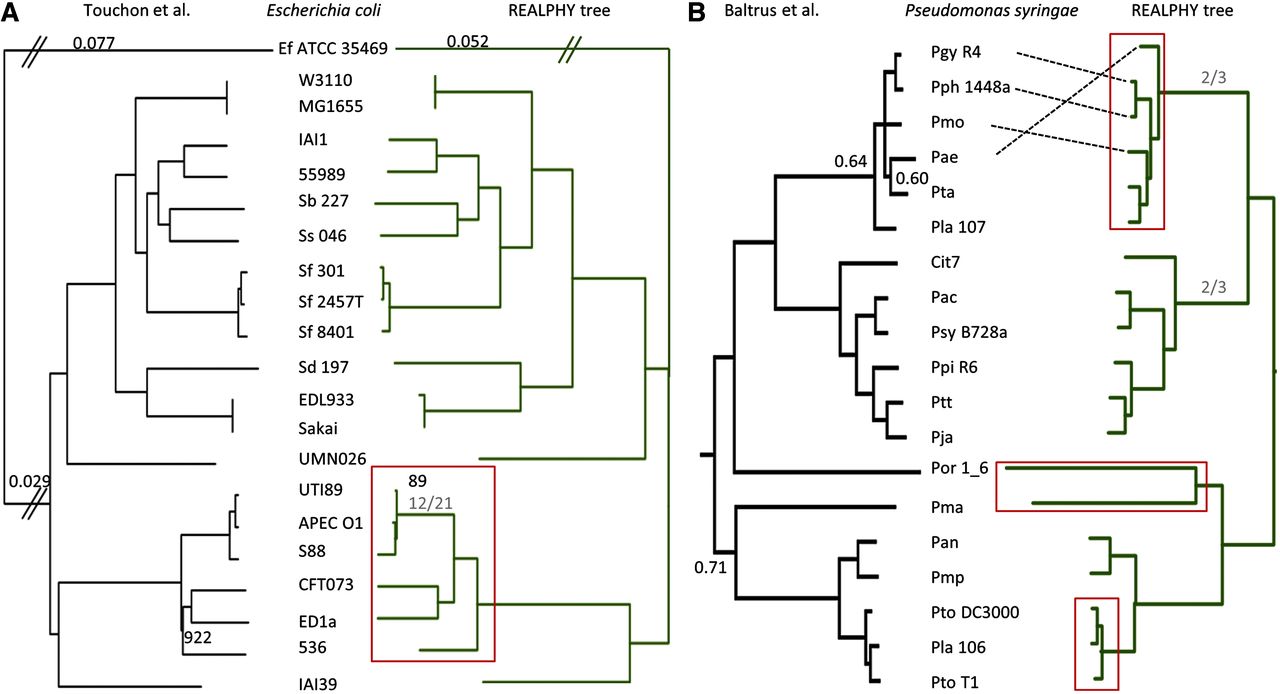
Studies of microbial evolutionary dynamics are being transformed by the availability of affordable high-throughput sequencing technologies, which allow whole-genome sequencing of hundreds of related taxa in a single study. Reconstructing a phylogenetic tree of these taxa is generally a crucial step in any evolutionary analysis. Instead of constructing genome assemblies for all taxa, annotating these assemblies, and aligning orthologous genes, many recent studies 1) directly map raw sequencing reads to a single reference sequence, 2) extract single nucleotide polymorphisms (SNPs), and 3) infer the phylogenetic tree using maximum likelihood methods from the aligned SNP positions. However, here we show that, when using such methods to reconstruct phylogenies from sets of simulated sequences, both the exclusion of nonpolymorphic positions and the alignment to a single reference genome, introduce systematic biases and errors in phylogeny reconstruction. To address these problems, we developed a new method that combines alignments from mappings to multiple reference sequences and show that this successfully removes biases from the reconstructed phylogenies. We implemented this method as a web server named REALPHY (Reference sequence Alignment-based Phylogeny builder), which fully automates phylogenetic reconstruction from raw sequencing reads.
Download PDF
Supplemental material
Short URL: https://tinyurl.com/y5yudnp8





![]()
![]()
![]()
![]()
Comments and Reactions
No comments yet. Why don't you write one?