SPA: A Probabilistic Algorithm for Spliced Alignment
Posted in Publications, publishedNo Comment
Erik van Nimwegen, Nicodeme Paul, Robert Sheridan and Mihaela Zavolan SPA: A Probabilistic Algorithm for Spliced Alignment. PLoS Genetics, 2006, 2(4), e24
ABSTRACT
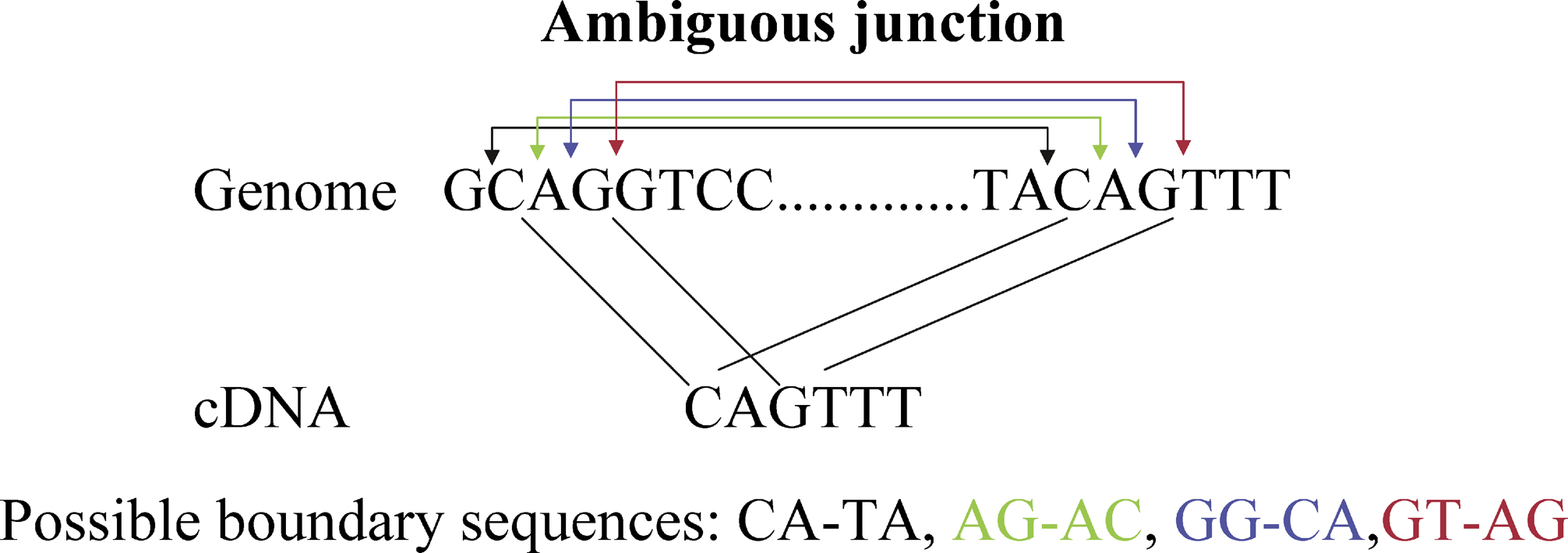
Recent large-scale cDNA sequencing efforts show that elaborate patterns of splice variation are responsible for much of the proteome diversity in higher eukaryotes. To obtain an accurate account of the repertoire of splice variants, and to gain insight into the mechanisms of alternative splicing, it is essential that cDNAs are very accurately mapped to their respective genomes. Currently available algorithms for cDNA-to-genome alignment do not reach the necessary level of accuracy because they use ad hoc scoring models that cannot correctly trade off the likelihoods of various sequencing errors against the probabilities of different gene structures. Here we develop a Bayesian probabilistic approach to cDNA-to-genome alignment. Gene structures are assigned prior probabilities based on the lengths of their introns and exons, and based on the sequences at their splice boundaries. A likelihood model for sequencing errors takes into account the rates at which misincorporation, as well as insertions and deletions of different lengths, occurs during sequencing. The parameters of both the prior and likelihood model can be automatically estimated from a set of cDNAs, thus enabling our method to adapt itself to different organisms and experimental procedures. We implemented our method in a fast cDNA-to-genome alignment program, SPA, and applied it to the FANTOM3 dataset of over 100,000 full-length mouse cDNAs and a dataset of over 20,000 full-length human cDNAs. Comparison with the results of four other mapping programs shows that SPA produces alignments of significantly higher quality. In particular, the quality of the SPA alignments near splice boundaries and SPA’s mapping of the 5′ and 3′ ends of the cDNAs are highly improved, allowing for more accurate identification of transcript starts and ends, and accurate identification of subtle splice variations. Finally, our splice boundary analysis on the human dataset suggests the existence of a novel non-canonical splice site that we also find in the mouse dataset. The SPA software package is available at http://www.biozentrum.unibas.ch/personal/nimwegen/cgi-bin/spa.cgi.
Short URL: https://tinyurl.com/yxfcqw9b





![]()
![]()
![]()
![]()
Comments and Reactions
No comments yet. Why don't you write one?